自體免疫疾病是我國十大重大傷病第三名,也是門診第三常見疾病。它是一種人體內的免疫系統異常活化而攻擊自己身體正常細胞的疾病,例如全身性紅斑狼瘡、類風濕關節炎、僵直性脊椎炎等。自體免疫疾病往往難以早期診斷出來,因而延遲了治療時機。患者飽受病痛之苦,必須終生治療與用藥,承受藥物引發的副作用,因此自體免疫疾病的早期診斷與治療是醫界長久以來的挑戰。
臨床上很難及早診斷症狀輕微的全身性紅斑狼瘡(SLE),並且發現SLE患者多為女性(占9/10),病患家族中也常發現不只一位家人罹患SLE。國衛院免疫醫學中心譚澤華特聘研究員與莊懷佳助研究員團隊,與台中榮總副院長藍忠亮醫師(現中國醫副院長)、高醫大顏正賢前院長組成合作研究團隊,以及台中榮總免疫風濕科洪維廷醫師、陳一銘醫師共同研究,證明MAP4K3 (又名GLK)蛋白激酶基因變異為SLE關鍵致病因子。此項研究已在今(2021)年10月發表於全球知名、風濕病醫學領域研究型第一名期刊《Annals of the Rheumatic Diseases》。
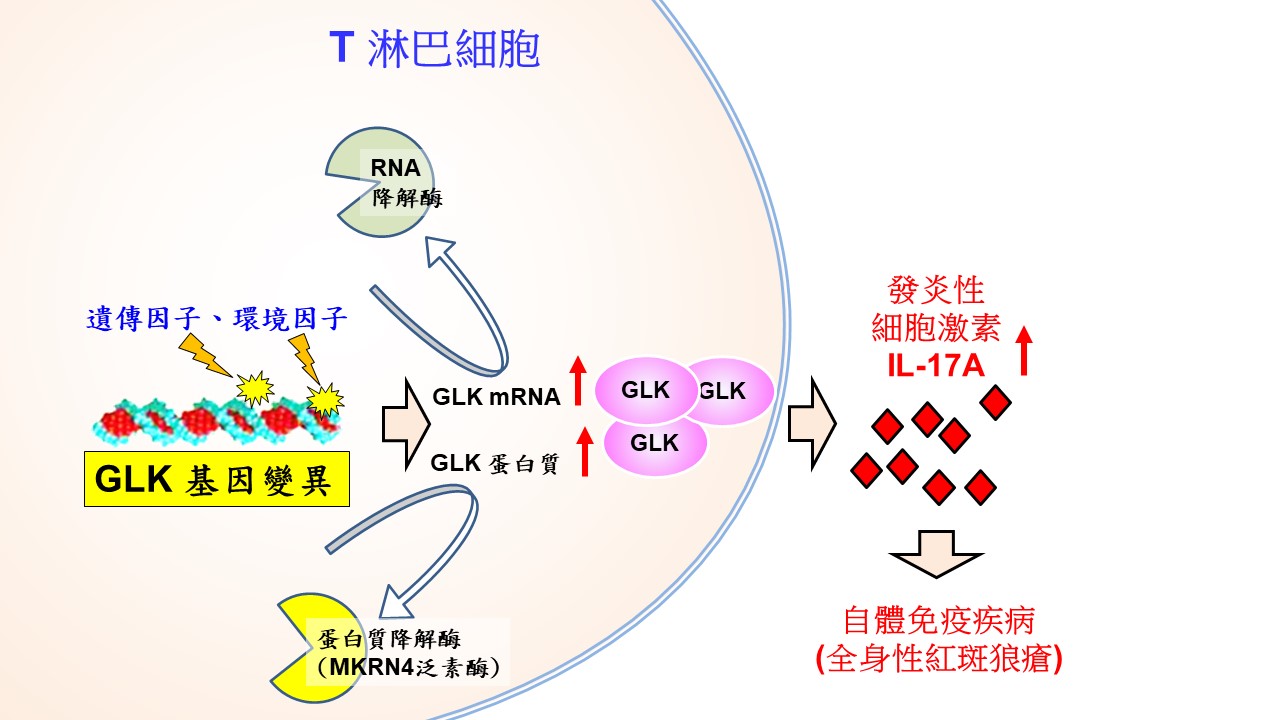
國衛院譚澤華特聘研究員率合作團隊,自2009年起研究SLE致病成因與精準之生物診斷標記,分析臨床檢體及基因改造小鼠研究,在2011年首度發現調控發炎反應的酵素蛋白激酶GLK是自體免疫疾病SLE的致病關鍵。再歷經7年研究,證實GLK過量表現在SLE病患之T淋巴細胞中,誘發IL-17A細胞激素大量產生,是造成許多自體免疫疾病的元凶。然而,GLK過量表現於SLE病患中的成因,多年來仍懸而未解。
為了增進統計分析之信賴度及研究結果之廣效性,研究團隊結合了中榮及高醫兩端的SLE病患檢體,以深度次世代基因定序(30萬reads/ gene)分析健康者、SLE病患、病患家屬之DNA樣本共431例。研究發現高達四成的SLE病患帶有GLK基因之體細胞性基因變異(somatic variant)或遺傳性基因變異(germline variant),這些基因變異造成GLK之mRNA或是蛋白質過度穩定而增量,因而誘發IL-17A細胞激素產生,導致自體免疫疾病。若能定期追蹤帶有GLK基因變異的女性家族成員,可望藉由基因檢測篩檢,提早在輕症時即診斷出SLE,以利及早治療。
除了鑑定出SLE疾病相關之基因變異,國衛院免疫醫學中心團隊乃全球首度為MKRN4基因驗明正身,推翻科學界既往認定MKRN4為「假基因」(pseudogene)的概念。研究證明MKRN4實為帶有重要功能之泛素酶,MKRN4泛素酶負責將過多之GLK泛素化且引發GLK降解而減量。然而帶有特定的GLK基因變異時,則會逃脫MKRN4調控之GLK降解作用,造成GLK在T淋巴球中過量表現,誘發IL-17A導致之自體免疫反應。
目前國際上研發的治療自體免疫疾病療法著重於生物製劑,如:抗IL-17A抗體目的在抑制細胞外過量的IL-17A,然而抗體藥發展成本較高。譚澤華特聘研究員研究團隊已開發可精準鎖定GLK之小分子抑制劑,直接從源頭關掉使IL-17A無法產生,具有潛力發展為成本較低的自體免疫疾病小分子標靶藥物,期待進一步推展臨床試驗。
此次多機構長年不懈的合作研究,帶來高度臨床應用價值的發現, GLK基因深度定序期待推動成為全身性紅斑狼瘡或是其他自體免疫疾病的精準個人化醫療基因檢測項目,嘉惠自體免疫疾病高風險族群。
研究論文全文:https://ard.bmj.com/content/early/2021/10/05/annrheumdis-2021-221010